



By: Jay Evans, USDA Beltsville Bee Lab
Knowing the complete genome of an animal, whether human, bee, or worm, can go a long way toward predicting how that animal looks, grows, and behaves. But to really know the source of interesting traits, one must study which parts of that genome are active at key times. Genes that provide the blueprints for individual proteins (a big part of the ‘active’ genome) are silent much of the time. When triggered, they spin off messenger RNAs, whose message is then translated into strings of amino acids that make up unique proteins. This triggering, or ‘transcription’, into messenger RNAs can be precise to individual genes or can link tens or hundreds of genes that turn on and off in a coordinated way. Coordinating the arrival of many proteins at once is essential for complex events such as building a limb, enacting an immune response, building a bank for memories, and numerous other needs of even the simplest organisms.
Humans have about 21,000 protein-coding genes and bees have maybe 16,000. These are depressingly average counts for mammals and insects, respectively, and far less than, say, earthworms and potatoes. Our exceptionalism, then, must reflect how well we use the genes we have. This relates directly to how well our cells turn particular genes on and off. When I first studied genetics, we were not given the whole story. Genes were turned on by promoters that hit a cartoonish landing pad on the chromosome just in front of the gene – and protein-building ensued. This view was nuanced a bit by the study of transcription factors that matched to varying degrees sites ‘near’ key genes, turning them all on at once. Science fans will know that anything with ‘factor’ in its name still has a bit of mystique (e.g., ‘virulence’, ‘queen’, and ‘fudge’ factors). Transcription factors are now well studied, however, and are validated as good predictors of which proteins need to work together during particular events in the lives of cells.
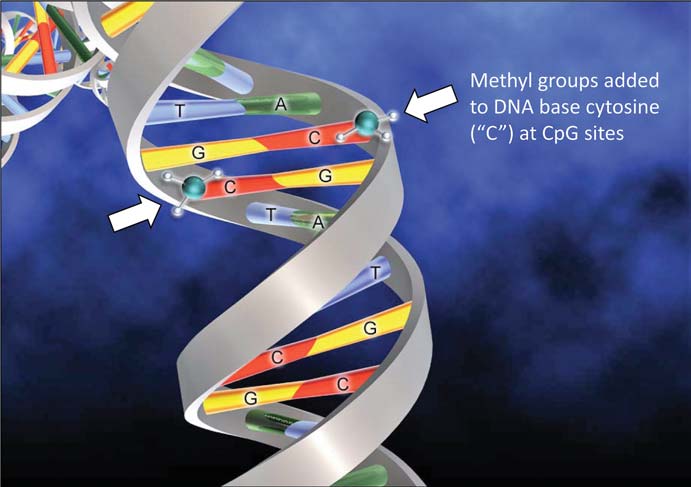
Many decades ago, discussion began on still more mysterious epigenetic (‘above the genome’) factors that regulated the activity of larger chromosome regions. Generally, epigenetic controls squash gene activity. One prime example for organisms with sex chromosomes (humans, yes, bees, no) involves the regulation of genes found on the chromosome shared by both sexes (the ‘X’ chromosome in our case). For many genes, it is healthier if their protein levels are roughly equal in males and females. Since females have two X chromosomes in their cells, there has to be some sort of dosage compensation for the proteins not related to sex differences, so they play well with proteins from the rest of the genome. To do this, genes encoding proteins on the X chromosome in males might be hyperactive to mimic those from XX females – but in reality, it is mostly the female X-encoded genes that slow down to match the boys. In fact, a good fraction of X chromosomes in female mammals is silenced by a process called methylation, an enzyme-driven swap of DNA components that makes these components less likely to be turned on. For you science buffs who remember the four bases of DNA, it is generally the cytosines that get dinged up in this way, neat by itself but also a perfect landmark for scientists to predict which parts of the genome are silenced.
Other than hearing these stories of dosage compensation by a somewhat sloppy brush, I admit to not really thinking much about methylation – UNTIL some fascinating work by my friend and hero, Professor Ryszard Maleszka from the Australian National University. Prof. Maleszka toiled for years as a leader in the sequencing of the honey bee genome. When it all came together, he was almost giddy to find that the bee genome had a full set of enzymes needed to tag specific chromosome segments with methylation and, importantly, maintain the faithfulness of that methylation tag over time. Soon, he and his team showed that these epigenetic forces were important in one of the most beautiful processes in bee biology, the generation of queen bees.
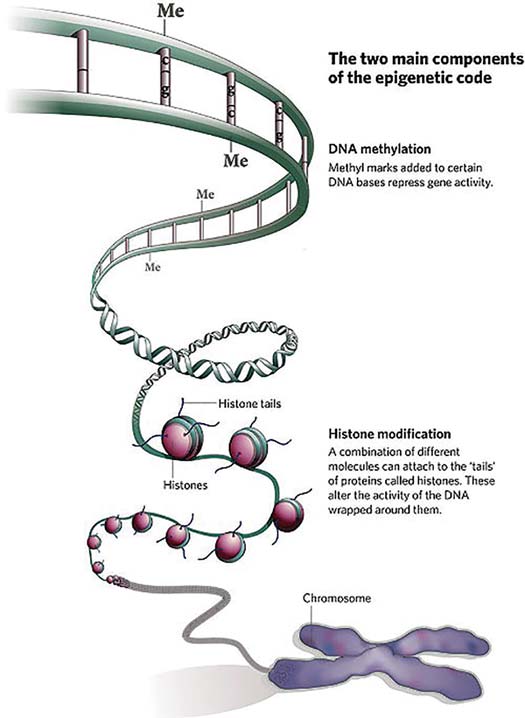
Queens and workers are indistinguishable at the gene level, a fact known by queen breeders who can graft without discrimination from female larvae to get their future queens. What sets them apart is the coordinated production of caste-biased proteins that, in the case of queen-destined larvae, speed metabolism and development and set the seeds for prodigious ovaries. Worker-destined larvae grow more slowly, and have different brain structures and barbed stingers, among many other differences. While the environmental causes of this split involve larval diet (royal jelly and its queen ‘factors’), inside each bee the queen path is set by the epic and apparently epigenetic regulation of hundreds of genes tuned to either a queen or worker fate. In a highly cited paper from 2008, Dr. Maleszka, his scientist wife Joanna, and their team produced clear evidence that silencing by methylation was key for this process. The paper, “Nutritional control of reproductive status in honeybees via DNA methylation” is freely available from the journal Science (https://science.sciencemag.org/content/319/5871/1827). Methylation is an imprecise paintbrush, more Monet than Michelangelo, but a critical gene showed shifts in methylation that reflected the queen or worker pathway. Interestingly, worker-destined larvae apparently had a higher rate of methylation for caste-related genes and the proof for the whole story came when the ANU team silenced a key enzyme that drives methylation and successfully pushed more larvae down the queen road, irrespective of diet!
Science moves on and additional epigenetic processes have been discovered since this work, but the message that whole cohorts of caste-biased genes can be tagged in a lasting way to impact their activity has held up beautifully. For a really recent take on it all, Professor Maleszka’s group recently teamed up with Marek Wojciechowski, Paul Hurd, and colleagues from England in a fascinating and freely available paper titled “Phenotypically distinct female castes in honey bees are defined by alternative chromatin states during larval development” (https://genome.cshlp.org/content/28/10/1532). These epigenetic changes are undeniable and the signals they leave behind have helped to identify numerous genes products tied to key traits like queen production, disease responses and even stinging tendencies. I will follow up with reviews of those traits in the future, and show how this discovery has been franchised over the past decade to help explain traits like queen production, or perhaps reproduction generally, in species far beyond honey bees. As a teaser, I mentioned that these methylation-related enzymes can repaint the same site even after cells divide. It turns out they ensure that some methylation patterns are faithful from drones or queens to their offspring, presenting great possibilities for a parent’s environment to shape the ways their offspring preps for life. Think ‘you are what your parents ate’, or suffered through, etc. There are also indications that some offspring traits more strongly reflect their dad’s versus mom’s contribution, a tendency that might either strengthen or challenge colony life. Stay tuned, it will be epigenetic.


